Molecular Interfaces
Molecular interfaces are ubiquitous and perform critical functions in organic electronic and photovoltaic devices. In typical organic solar cells sunlight is harvested by organic species and charge separation is driven by the potential energy difference at an interface between donor and acceptor species. In any organic device, charge transport to external circuits depends on interfaces between active molecular layers and electrode materials. The properties and functionality of these critical interfaces cannot be deduced directly from those of their isolated constituents. Rather, they emerge from quantum mechanical interactions at the atomistic scale. Predicting the properties of molecular interfaces thus requires a fully quantum mechanical first principles approach.
The configuration space of molecular interfaces is infinitely vast, owing to the endless possibilities of combining layers of one or more molecular species with different substrates. Layers of the same molecular species may adopt different structures on different substrates and therefore exhibit different electronic and optical properties. Furthermore, epitaxial templating may enable stabilizing meta-stable crystal structures with desirable properties in thin film form. Efficient algorithms may significantly accelerate the discovery and design of molecular interfaces with enhanced properties. We are developing a first-principles framework for structure and property prediction of molecular interfaces.
Structure prediction of Epitaxial Organic Interfaces with Ogre
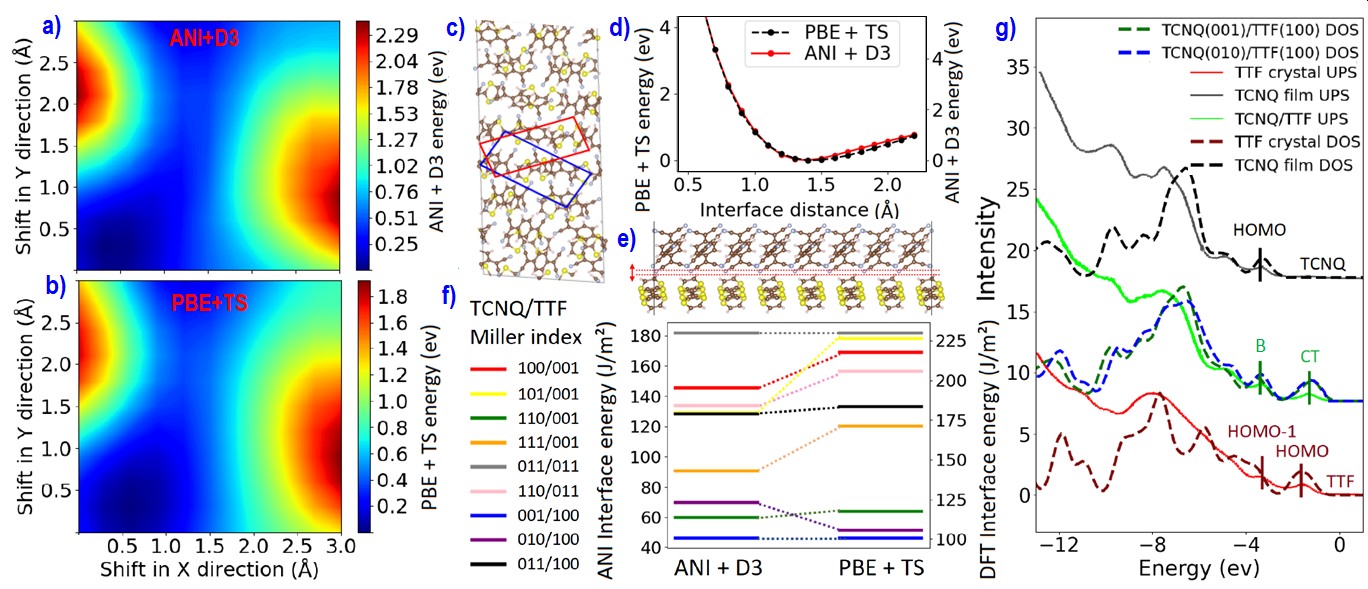
Highly ordered epitaxial interfaces between organic semiconductors are considered as a promising avenue for enhancing the performance of organic electronic devices including solar cells and transistors, thanks to their well-controlled, uniform electronic properties and high charge carrier mobilities. The electronic structure of epitaxial organic interfaces, and hence their functionality in devices, are inextricably linked to their structure. We have implemented in the Ogre code, a method for structure prediction of epitaxial organic interfaces based on lattice matching followed by surface matching. The lattice matching step produces domain-matched interfaces, where commensurability is achieved with different integer multiples of the substrate and film unit cells. In the surface matching step, Bayesian optimization (BO) is used to find the interfacial distance and registry between the substrate and film. The BO objective function is based on dispersion corrected deep neural network interatomic potentials. These are shown to be in qualitative agreement with density functional theory (DFT) regarding the optimal position of the film on top of the substrate (panels a-e) and the ranking of putative interface structures (panel f). Ogre is used to investigate the epitaxial interface of the acceptor 7,7,8,8-tetracyanoquinodimethane (TCNQ) grown on top of the donor tetrathiafulvalene (TTF). The electronic structure of the TCNQ/TTF interface has been probed by ultraviolet photoemission spectroscopy (UPS), but its structure has been hitherto unknown [Organic Electronics 48, 371 (2017)]. We find that TCNQ(001) on top of TTF(100) is the most stable interface configuration, closely followed by TCNQ(010) on top of TTF(100) (panel f). The density of states, calculated using DFT, is in excellent agreement with UPS experiments, including the presence of an interface charge transfer state (panel g).
J. Phys. Chem. C, DOI: 10.1021/acs.jpcc.3c02384 (2023)
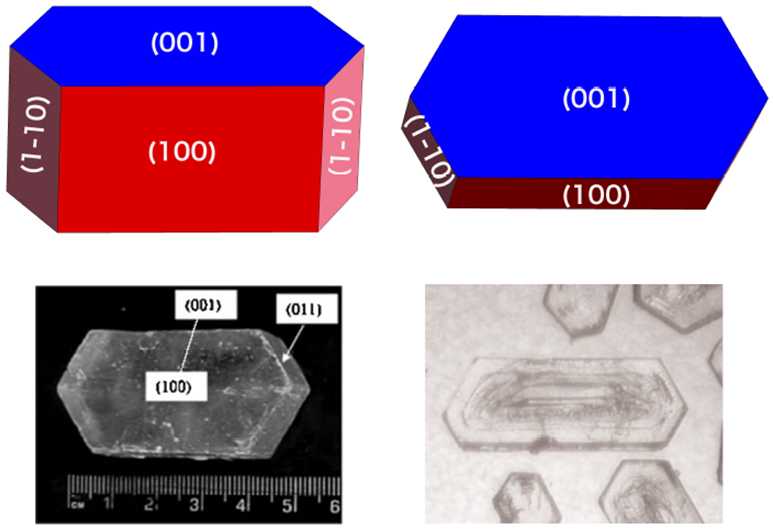
Ogre: A Python package for molecular crystal surface generation with applications to surface energy and crystal shape prediction
We have developed a new code, Ogre, for generating molecular crystal surface slabs, and the accompanying OgreSWAMP utilities for streamlining the calculation of surface energies and Wulff shapes. Ogre takes as input a bulk crystal structure and generates surface slab models with user-specified Miller indices, number of layers, and vacuum space. When cleaving molecular crystal surfaces, Ogre uses a graph representation to identify molecules and ensure the molecules at the surface remain intact. Ogre identifies all the non-equivalent surfaces based on the crystal’s space group symmetry and outputs slab structures with all possible terminations for each unique surface. These can be used to calculate the surface energies and predict the crystal shape. The Wulff shape of aspirin, produced by Ogre, is in good agreement with experimental data from Journal of Pharmaceutical Sciences 96, 2134 (2007) and Journal of Pharmaceutical Sciences 97, 1361 (2008).